
Machine Learning Approaches
We develop ML methods to overcome large phase-space challenge in chemically-complex materials.
The PREDICT Approach
PRedict properties from Existing Database in Complex materials Territory (PREDICT)

In the realm of high entropy materials where orders of magnitude compositions are possible, the conventional high-throughput DFT strategy for materials discovery fails miserably.
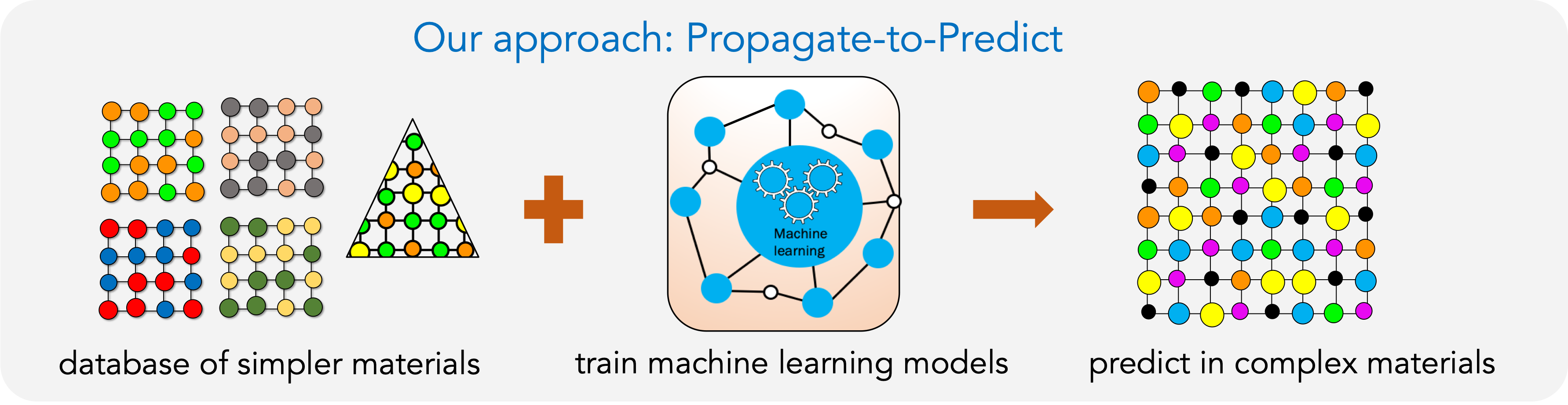
We have developed a new approach called PREDICT (PRedict properties from Existing Database in Complex materials Territory). A database of simpler/binary materials is prepared once-and-for-all from DFT calculations. It is used to train ML model that can then predict properties in complex multi-elemental materials containing different elements. We have used this approach to successfully predict various properties including vacancy formation and migration energies, vibrational entropies, stacking fault energies and elastic constants in multi-elemental alloys.
Figure shows a comparison of elastic constants between DFT vs ML in a 5-elemental alloy.

Charge-Density Image Recognition Approach
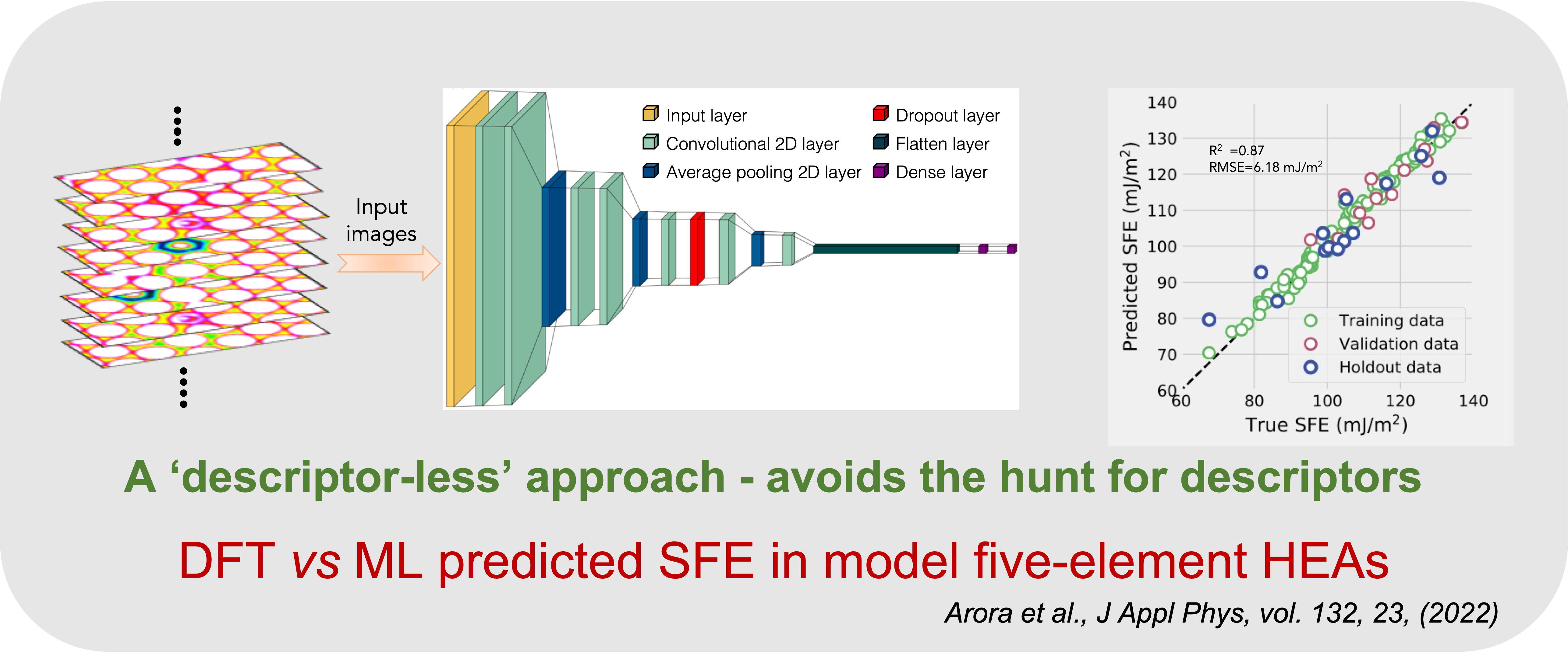
Descriptors are a means to incorporate physics in ML models. Ascertaining correct descriptors has become a vexing issue. We propose that charge density is the fundamental descriptor which captures electronic and atomic descriptions.Various descriptors such as atomic radius, electronegativity, charge transfer, valence electrons, etc. are implicitly included in charge density.
Hohenberg-Kohn theorem: Total energy of a many-body electron system is a functional of charge density.
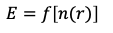

We have built a charge-density based convolutional neural network (CNN) model trained on charge density from DFT calculations. The model can predict stacking fault energies (SFE) in high entropy alloys (HEAs). A good comparison of DFT-calculated vs ML-predicted SFE is shown below.
Database and Codes
All our data including ML codes in Jupyter notebook are shared on Github.
Vibrational Entropy