
Atomistic Simulation Methods to Predict Protein-Surface Interactions
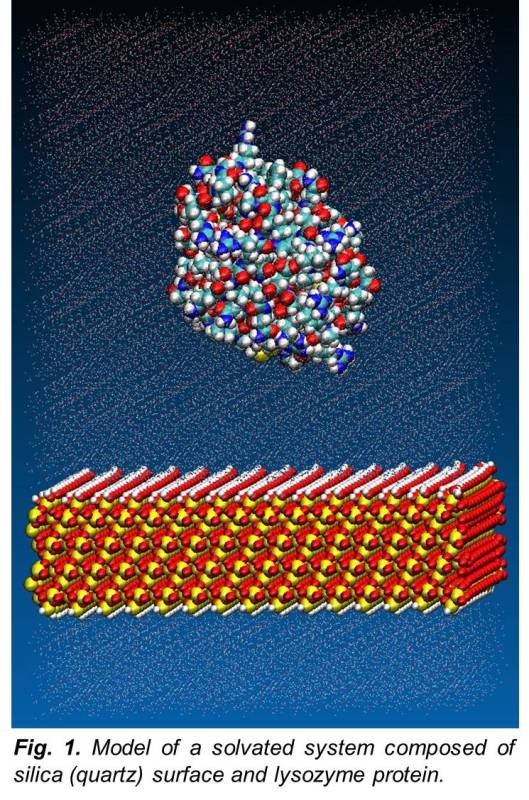 Past research conducted in this area was aimed at extending the results of earlier studies within the group that had the objective of generating a CHARMM dual-force-field suitable for simulation studies of peptide interactions on various self-assembled monolayer (SAM) surfaces. Those studies utilized small test-probe peptides to validate the force field by comparing the calculated free energy of adsorption (ΔGads) of each test probe peptide on various SAM surfaces with experimental results. The simulations utilized a biased energy replica-exchange molecular dynamics (REMD) to adequately sample configurational space of the peptide at specific distances of the peptide from the surface, called surface separation distance (SSD). Research then focused on extending those studies toward the goal of performing simulations of the interactions of larger proteins of more practical interest. Of particular interest was the interaction of proteins on surfaces such as silica glass, polymethylmethacrylate (PMMA), and high-density polyethylene (HDPE) as relevant model surfaces for biomaterials applications. Studies examined the free energy of adsorption of small test-probe peptides on these surfaces for the purpose of validating the force field for the simulation of the adsorption behavior of larger proteins on these surfaces. Simulations were then performed on lysozyme as a test case for larger proteins.
Past research conducted in this area was aimed at extending the results of earlier studies within the group that had the objective of generating a CHARMM dual-force-field suitable for simulation studies of peptide interactions on various self-assembled monolayer (SAM) surfaces. Those studies utilized small test-probe peptides to validate the force field by comparing the calculated free energy of adsorption (ΔGads) of each test probe peptide on various SAM surfaces with experimental results. The simulations utilized a biased energy replica-exchange molecular dynamics (REMD) to adequately sample configurational space of the peptide at specific distances of the peptide from the surface, called surface separation distance (SSD). Research then focused on extending those studies toward the goal of performing simulations of the interactions of larger proteins of more practical interest. Of particular interest was the interaction of proteins on surfaces such as silica glass, polymethylmethacrylate (PMMA), and high-density polyethylene (HDPE) as relevant model surfaces for biomaterials applications. Studies examined the free energy of adsorption of small test-probe peptides on these surfaces for the purpose of validating the force field for the simulation of the adsorption behavior of larger proteins on these surfaces. Simulations were then performed on lysozyme as a test case for larger proteins.
Defense Threat Reduction Agency (DTRA)
Grant number HDTRA1-10-1-0028 (PI: Latour)