
Molecular Dynamics Simulation - Peptide Surface Interactions
Poster 1 Poster 2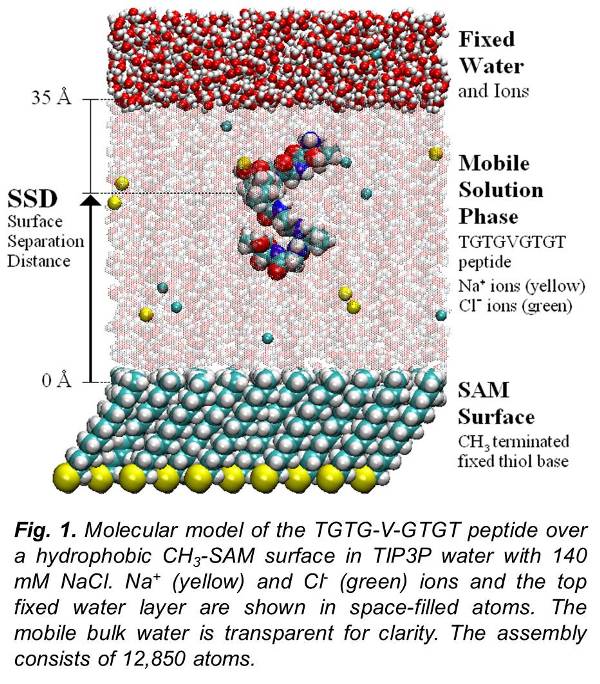 The study of protein-surface interaction is one of the crucial topics in the field of implants and biomaterials. Very little is understood regarding the molecular level events that control protein adsorption behavior. Understanding how proteins interact with these implant surface can help designing better biocompatible medical devices. With the exploitation of computers and algorithm, it is possible to study the adsorption process at the atomistic level. Recent advances in the field of Molecular dynamics (MD) can help develop an understanding of the behavior of these systems at the molecular level. MD studies employ a potential energy function in the form of force field to describe the various interactions and energies of the complex bio-molecular system. These force fields were developed for predicting the behavior of protein in solvent.
The study of protein-surface interaction is one of the crucial topics in the field of implants and biomaterials. Very little is understood regarding the molecular level events that control protein adsorption behavior. Understanding how proteins interact with these implant surface can help designing better biocompatible medical devices. With the exploitation of computers and algorithm, it is possible to study the adsorption process at the atomistic level. Recent advances in the field of Molecular dynamics (MD) can help develop an understanding of the behavior of these systems at the molecular level. MD studies employ a potential energy function in the form of force field to describe the various interactions and energies of the complex bio-molecular system. These force fields were developed for predicting the behavior of protein in solvent.
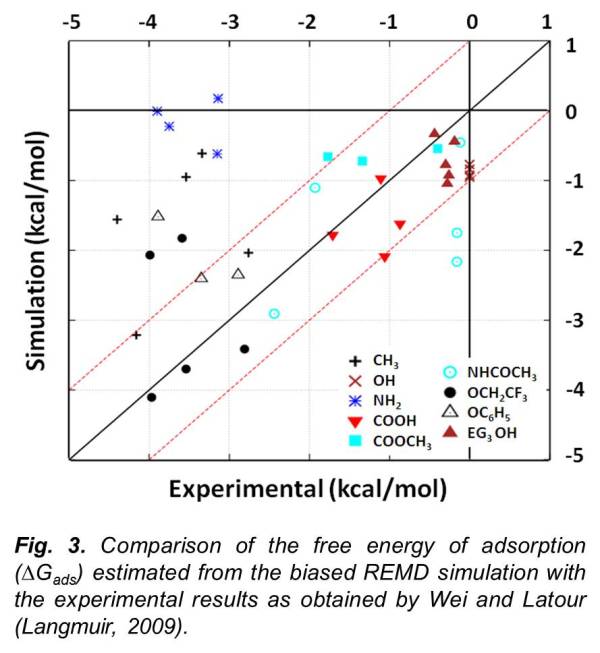
In order to evaluate the transferability of empirical force fields for all-atom molecular simulations of protein adsorption behavior, we developed and applied a method to calculate the adsorption free energy (ΔGads) of model peptides on functionalized surfaces for comparison with available experimental data. Simulations were conducted using the CHARMM program and force field using a host-guest peptide with the sequence TGTG-X-GTGT (where G and T are glycine and threonine amino acid residues, respectively, with X representing valine, threonine, aspartic acid, phenylalanine or lysine) over nine different functionalized alkanethiol self-assembled monolayer (SAM) surfaces with explicitly represented solvent (mTIP3P water model). ΔGads was calculated using biased-energy replica exchange molecular dynamics to compare with the experimental results. Although the CHARMM force-field was able to represent ΔGads within 1 kcal/mol of the experimental values for most systems, deviations as large as 4 kcal/mol were found for others.
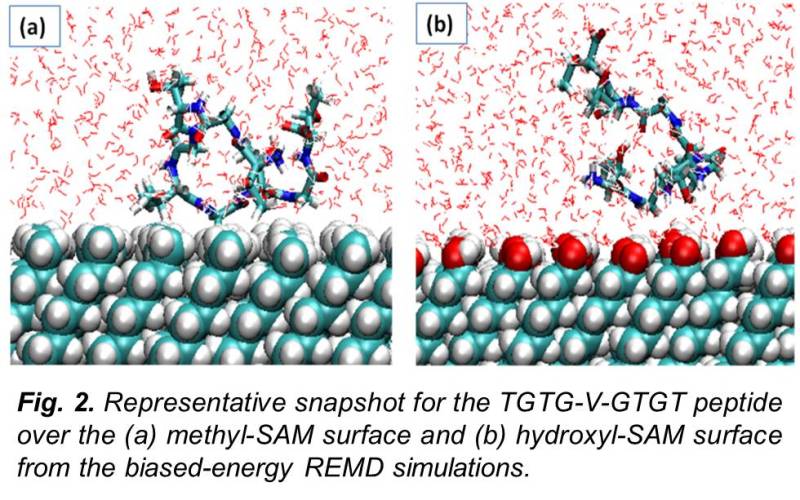
In particular, the simulations revealed that CHARMM underestimates the strength of adsorption on the hydrophobic and positively charged amine surfaces. These results provided a means for force field evaluation and modification for the development and validation of an interfacial force field for the accurate simulation of protein adsorption behavior. Based on these findings, we subsequently developed an interfacial force-field that could be utilized along with the CHARMM force-field in a "dual force-field simulation engine" in order to correct the deviations found in CHARMM force field simulation.
NIH/NIBIB, R01 grant no. EB006163 (PI: Latour)